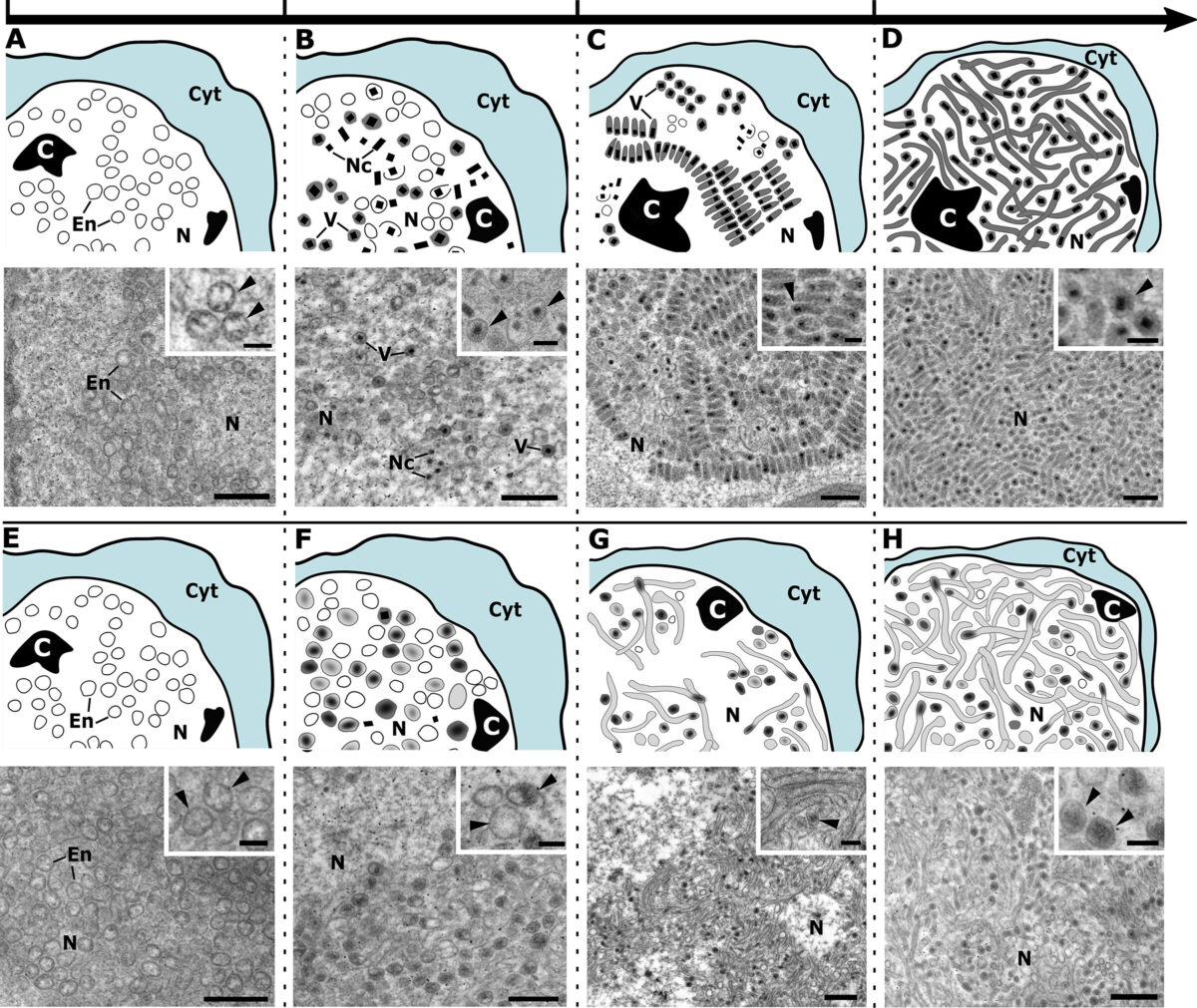
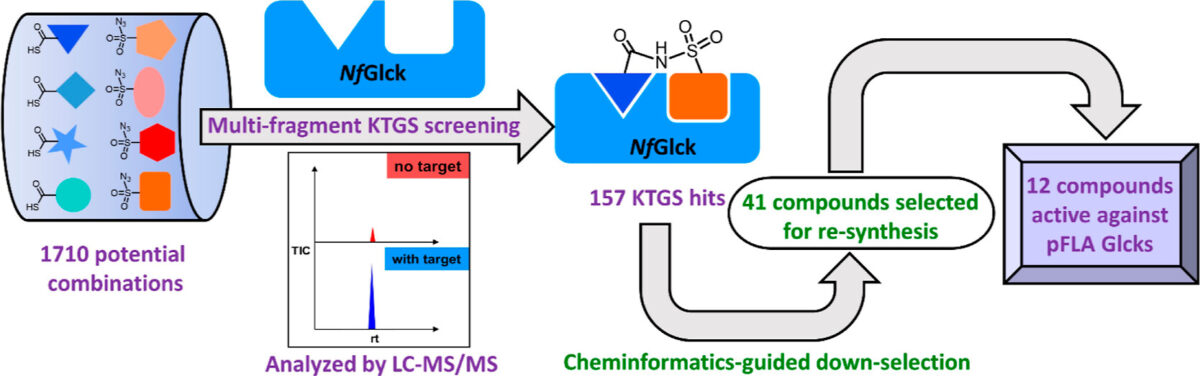
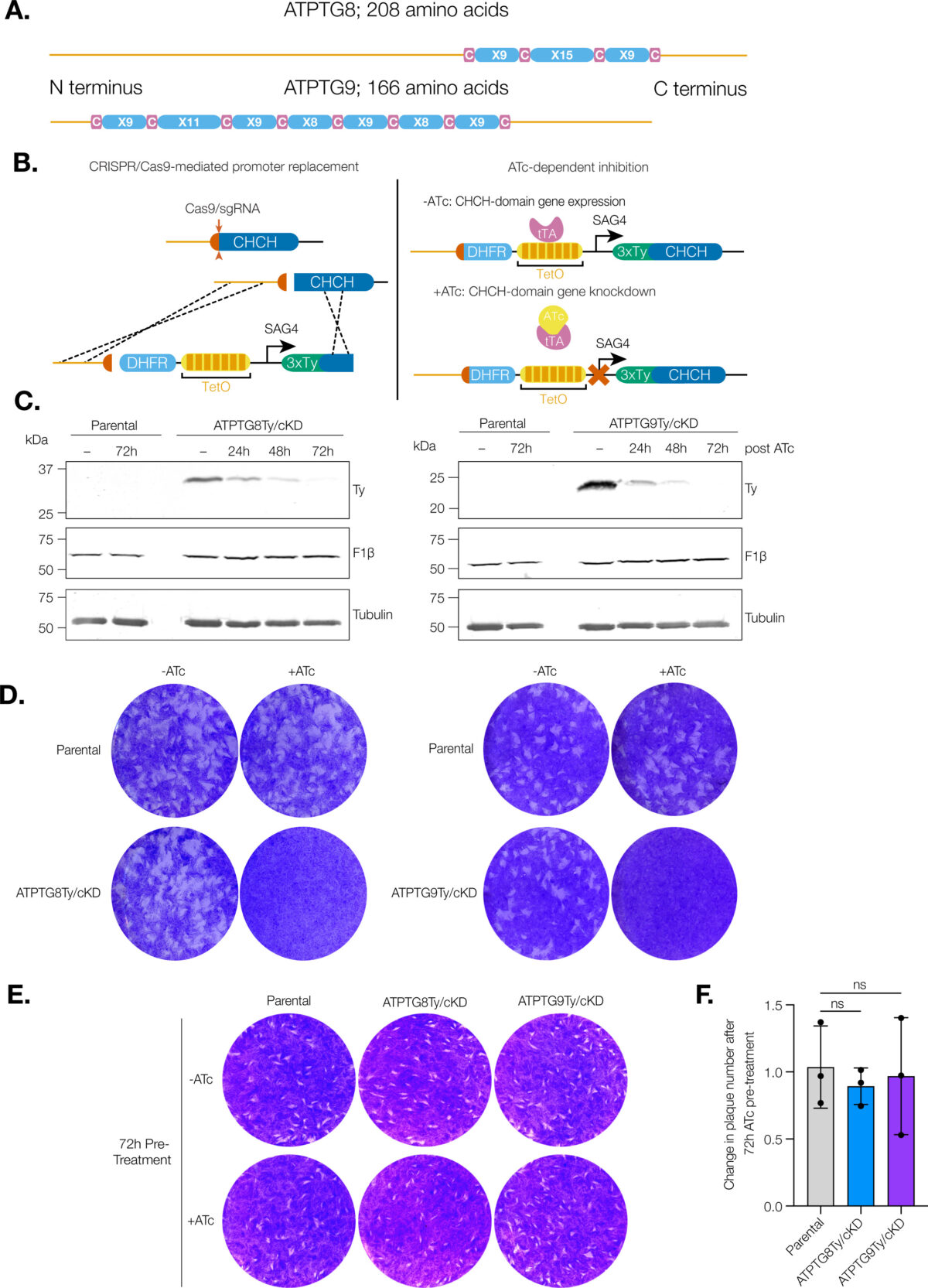
Undergraduate Research Experience Sparked Interest in Parasitology for Graduate Student

My name is Victoria Mendiola and I am a PhD candidate in Dennis Kyle's lab studying drug-induced dormancy in Plasmodium falciparum, the parasite responsible for malaria. I have been at UGA for four years but originally received my BSc in Biology and MSc of Integrative Biology from Kennesaw State University in Kennesaw, GA. My interest …