cGAS-STING Pathway Activation during Trypanosoma cruzi Infection Leads to Tissue-Dependent Parasite Control
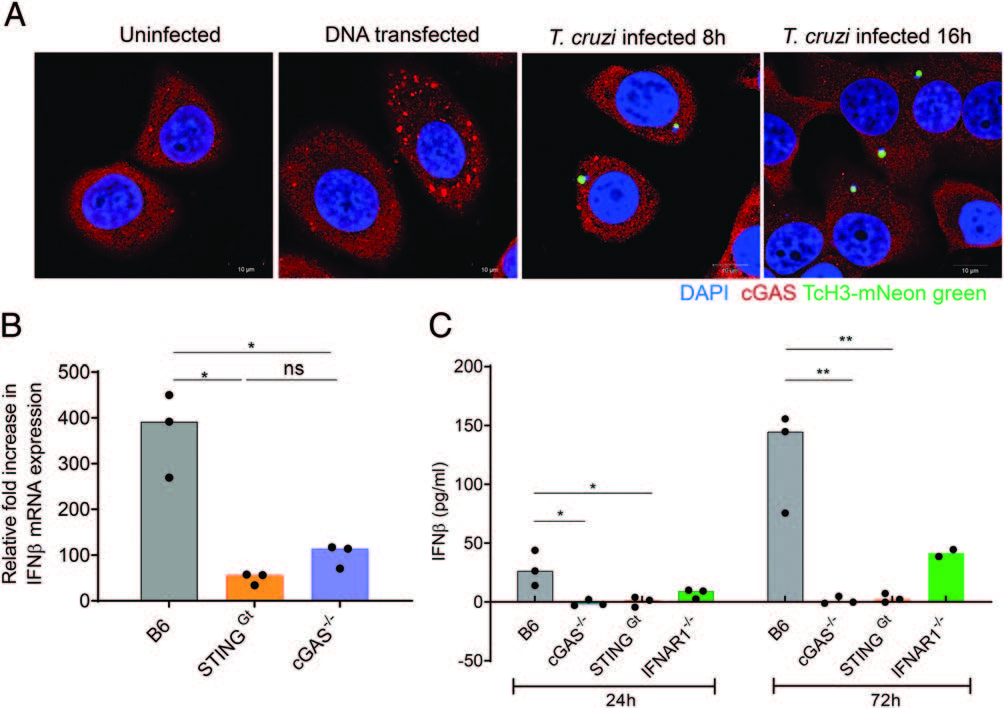
Host cell invasion by Trypanosoma cruzi is a markedly silent process, with limited host transcriptional changes indicative of innate immune recognition, except for a modest type I IFN (IFN-I) response. In this study, we show that T. cruzi-induced IFN-β production was nearly abolished in primary murine cGAS-/- or stimulator of IFN genes (STING)-deficient (STINGGt) macrophages and fibroblasts. T. cruzi infection did not impact the ability of IFN-regulatory factor reporter macrophages to respond to classical cGAS-STING agonists, indicating that the limited IFN-β induction is not due to active parasite suppression. cGAS-/-, STINGGt, and IFN-α/β receptor-/- (IFNAR-/-) macrophages infected with T. cruzi yielded significantly higher numbers of amastigotes compared with wild-type macrophages; however, the impact of the STING pathway during infection in vivo is more complex. Despite an initial increase in parasite growth, STINGGt and IFNAR-/- mice ultimately had lower parasite burden in footpads as compared with wild-type mice, demonstrating a role for IFN-I expression in potentiating parasite growth at the infection site. STING pathway activation had little impact on parasite levels in the skeletal muscle; however, in the heart, cGAS-/- and STINGGt mice, but not IFNAR-/- mice, accumulated higher acute parasite loads, suggesting a protective role of STING sensing of T. cruzi in this organ that was independent of IFN-I. Together, these results demonstrate that host cGAS-STING senses T. cruzi infection, enhancing parasite growth at the site of entry, and contributes to acute-phase parasite restriction in the heart, a major site of tissue damage in chronic T. cruzi infection.
Natasha Perumal, Brooke White, Fernando Sanchez-Valdez, Rick L Tarleton. J Immunol. 2023 Aug 21;ji2300373. doi: 10.4049/jimmunol.2300373.