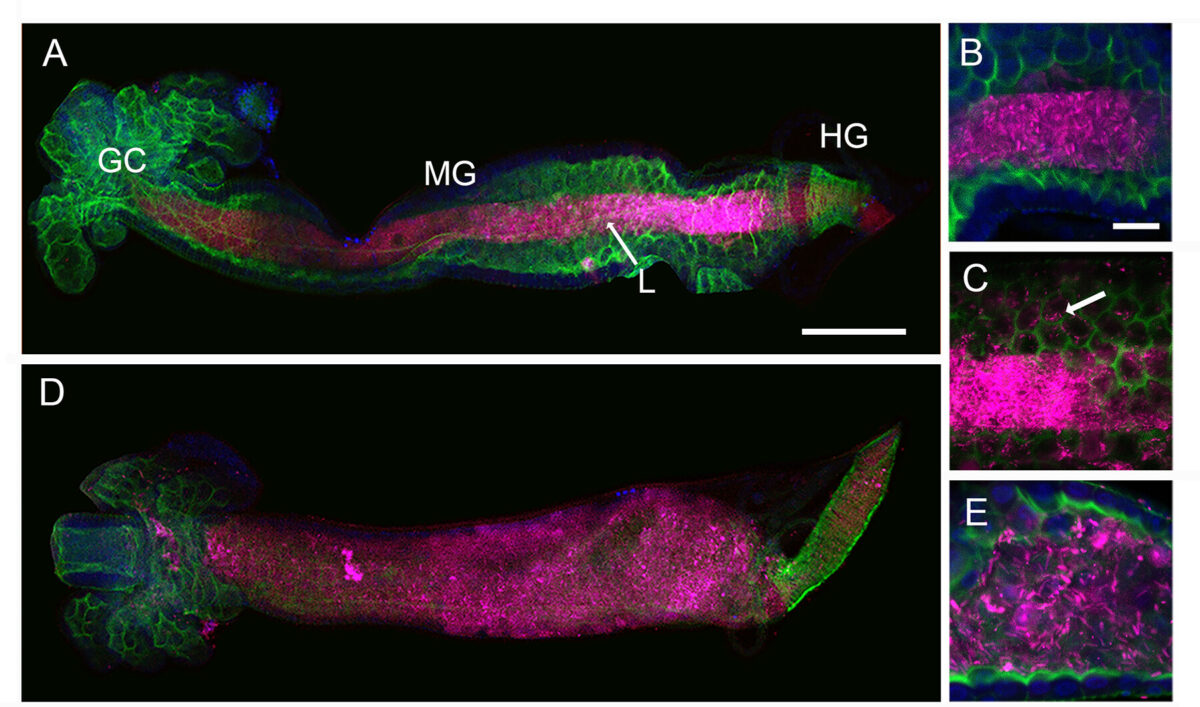
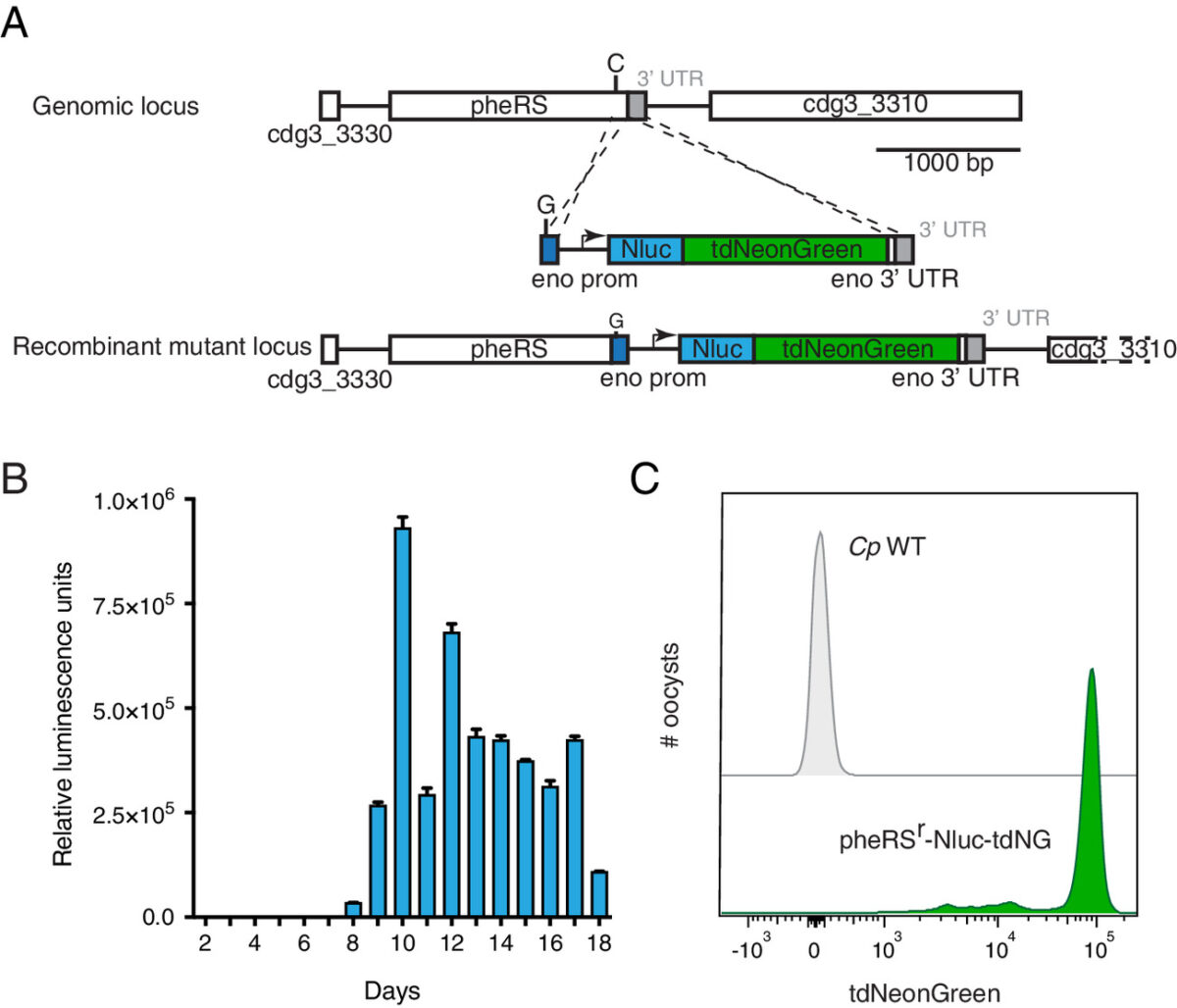
Genetic crosses within and between species of Cryptosporidium
Parasites and their hosts are engaged in reciprocal coevolution that balances competing mechanisms of virulence, resistance, and evasion. This often leads to host specificity, but genomic reassortment between different strains can enable parasites to jump host barriers and conquer new niches. In the apicomplexan parasite Cryptosporidium, genetic exchange has been hypothesized to play a prominent role …