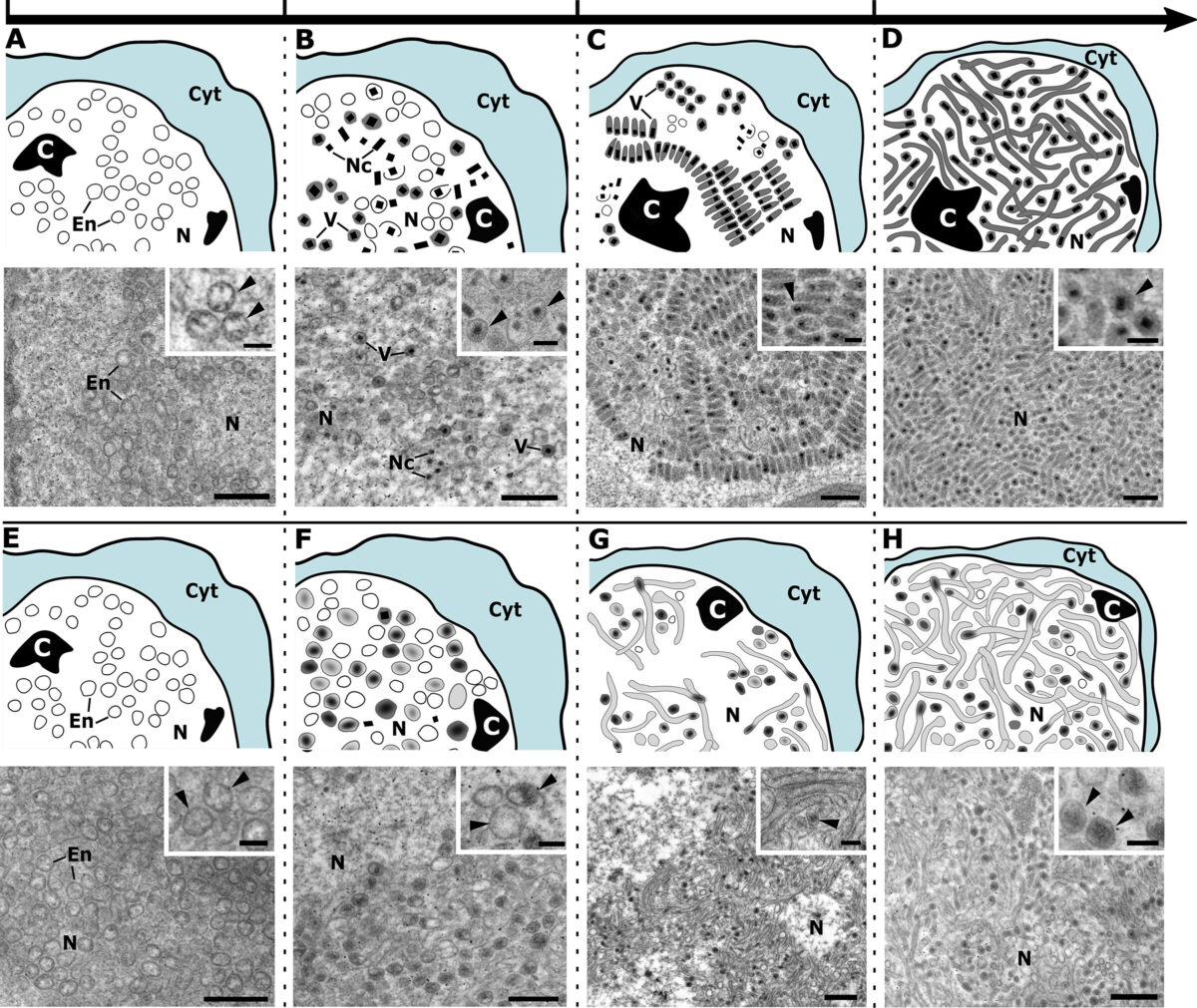
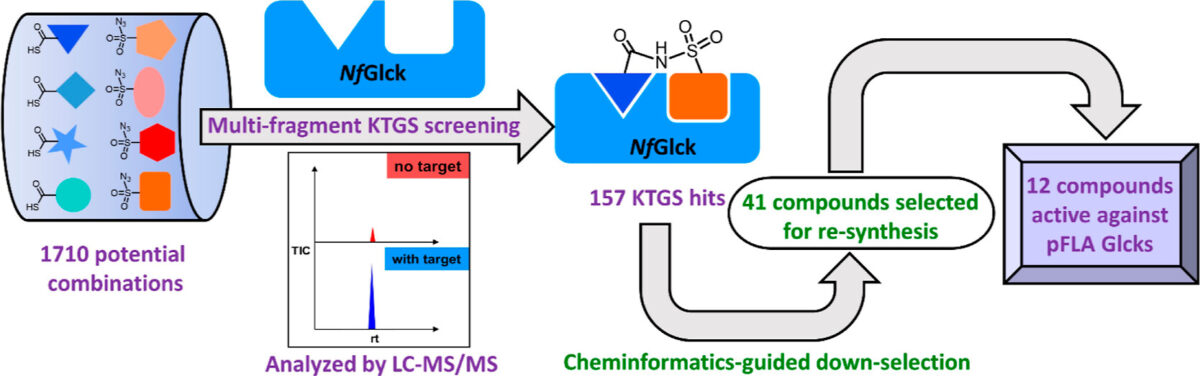
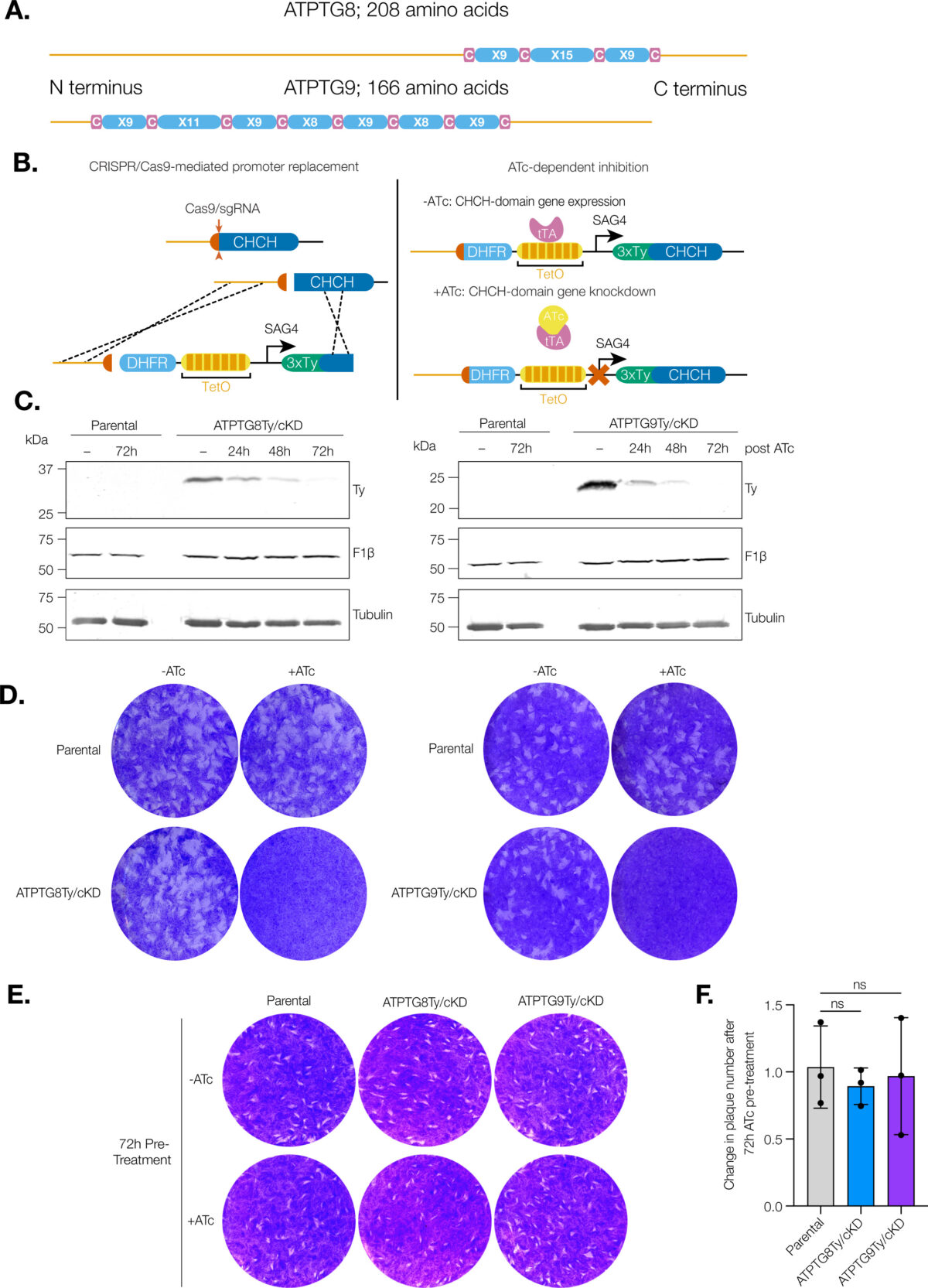
Blood meals from ‘dead-end’ vertebrate hosts enhance transmission potential of malaria-infected mosquitoes
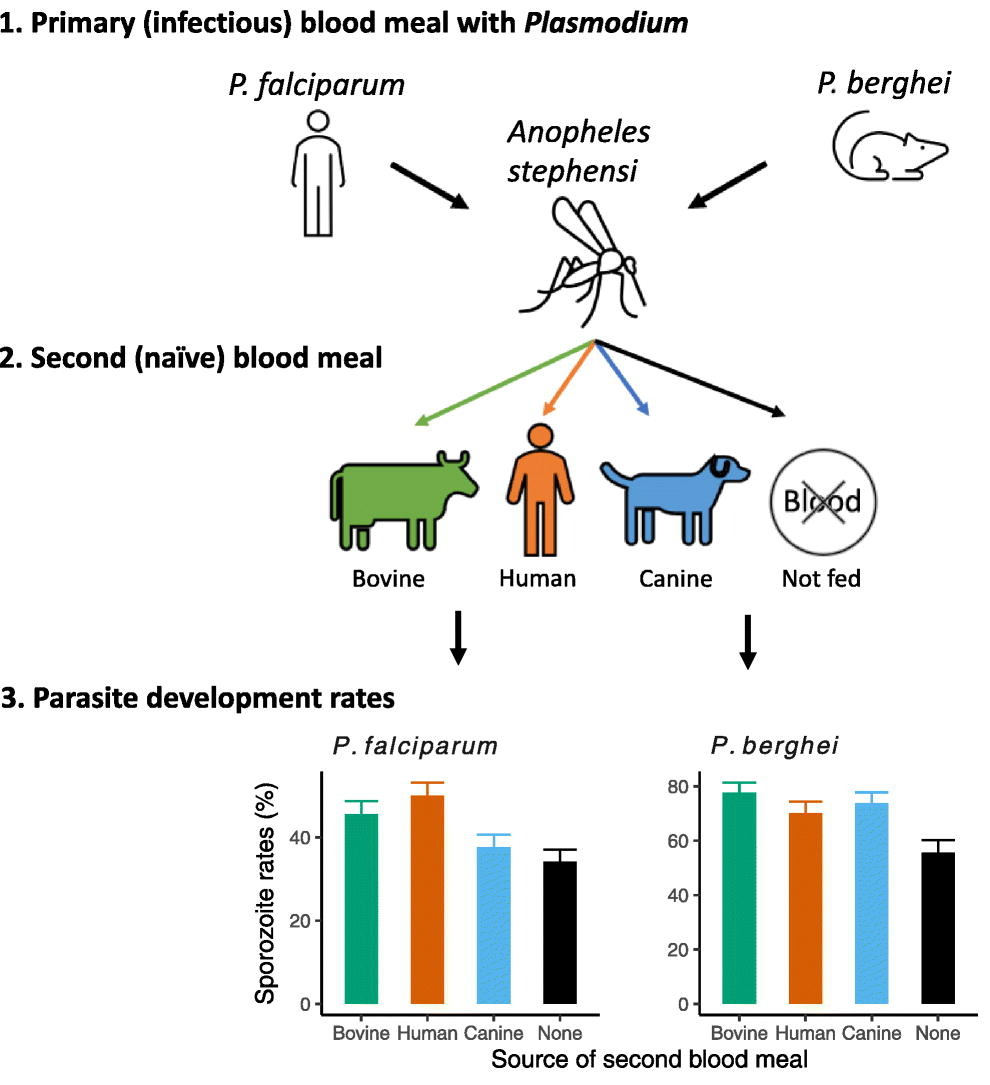
Ingestion of an additional blood meal(s) by a hematophagic insect can accelerate development of several vector-borne parasites and pathogens. Most studies, however, offer blood from the same vertebrate host species as the original challenge (for e.g., human for primary and additional blood meals). Here, we show a second blood meal from bovine and canine hosts …