A Krüppel-like factor is required for development and regeneration of germline and yolk cells from somatic stem cells in planarians
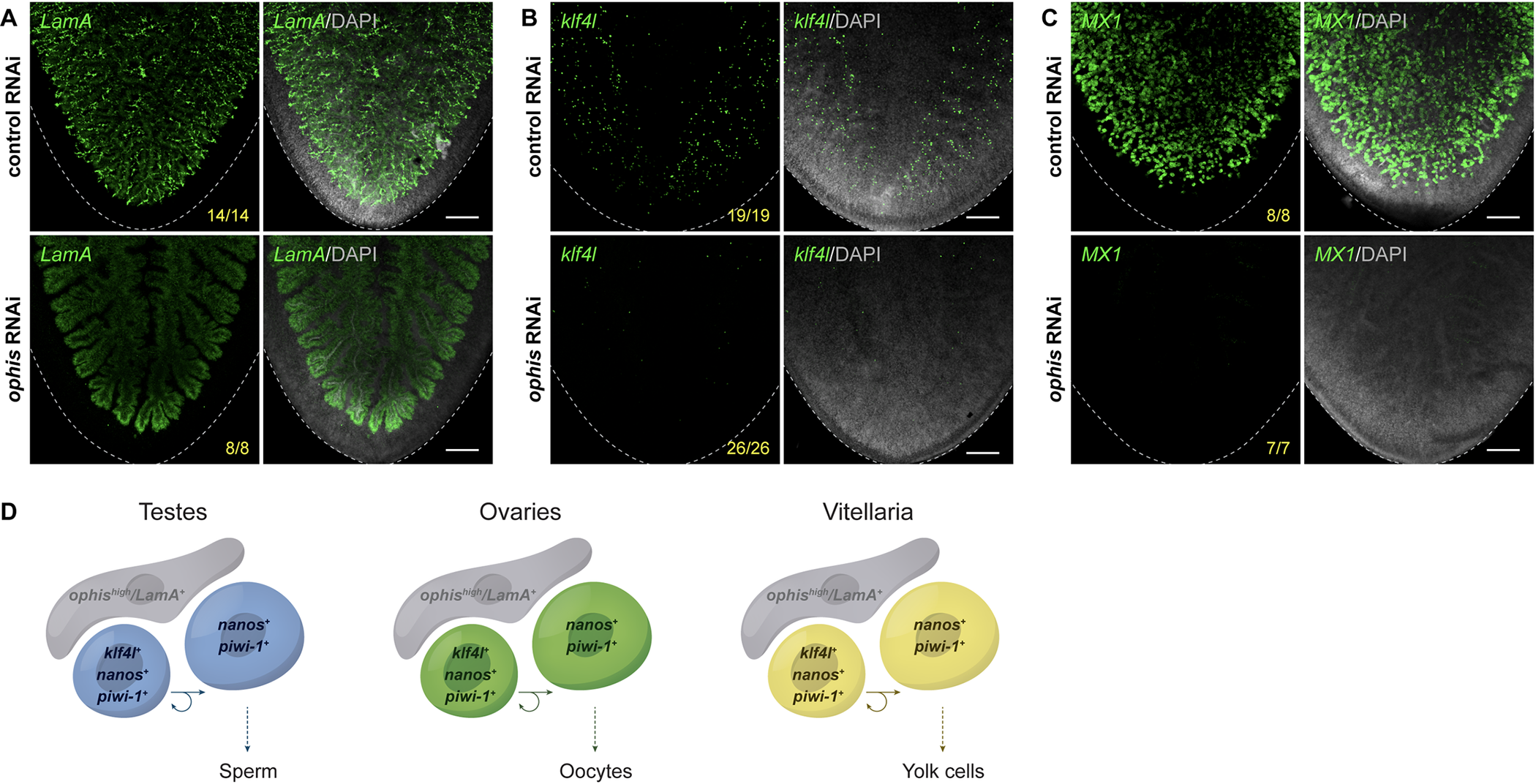
Fig 8. Germ cell niche factor ophis is required to sustain yolk cell production/vitellogenesis.(A–C) Maximum intensity projections of confocal sections showing FISH of LamA (A), klf4l (B), and MX1 (C) (green) in the ventral posterior region of sexually mature control versus ophis RNAi animals. Dashed line denotes planarian boundary. N = 3 to 5 experiments, …