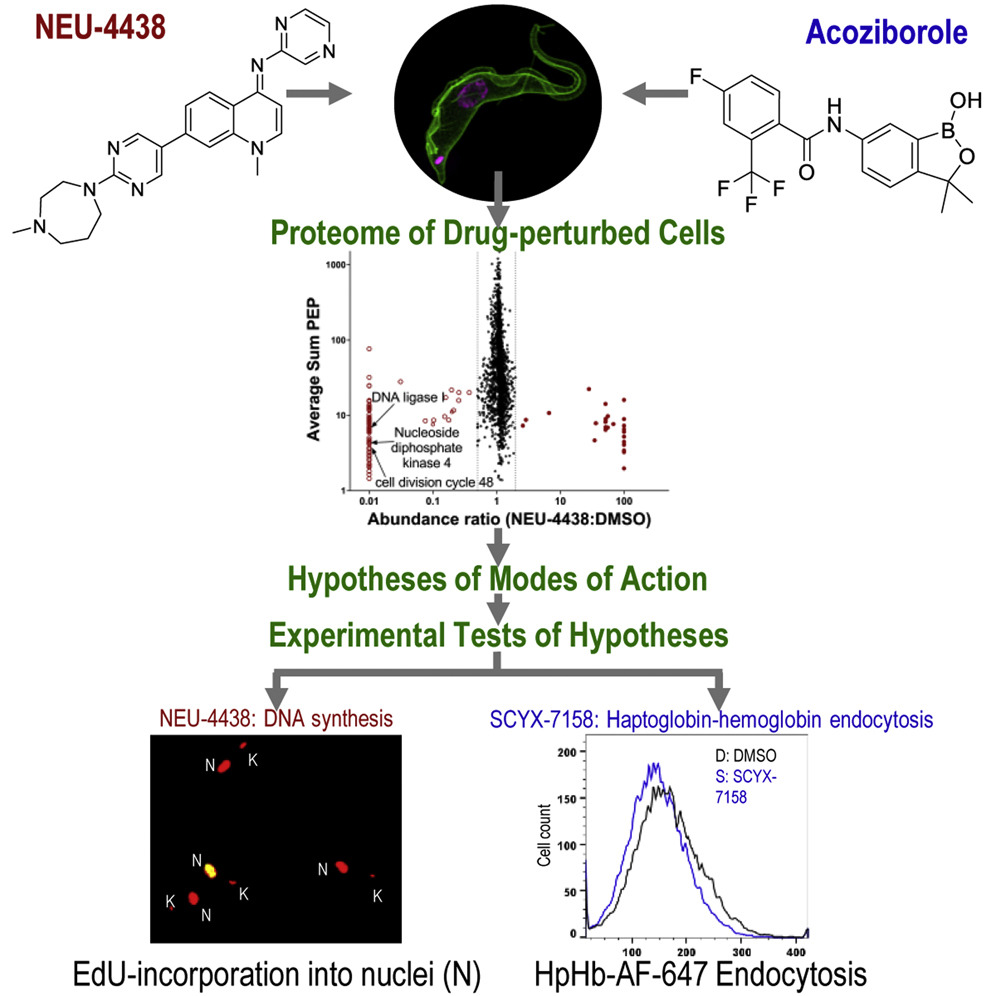
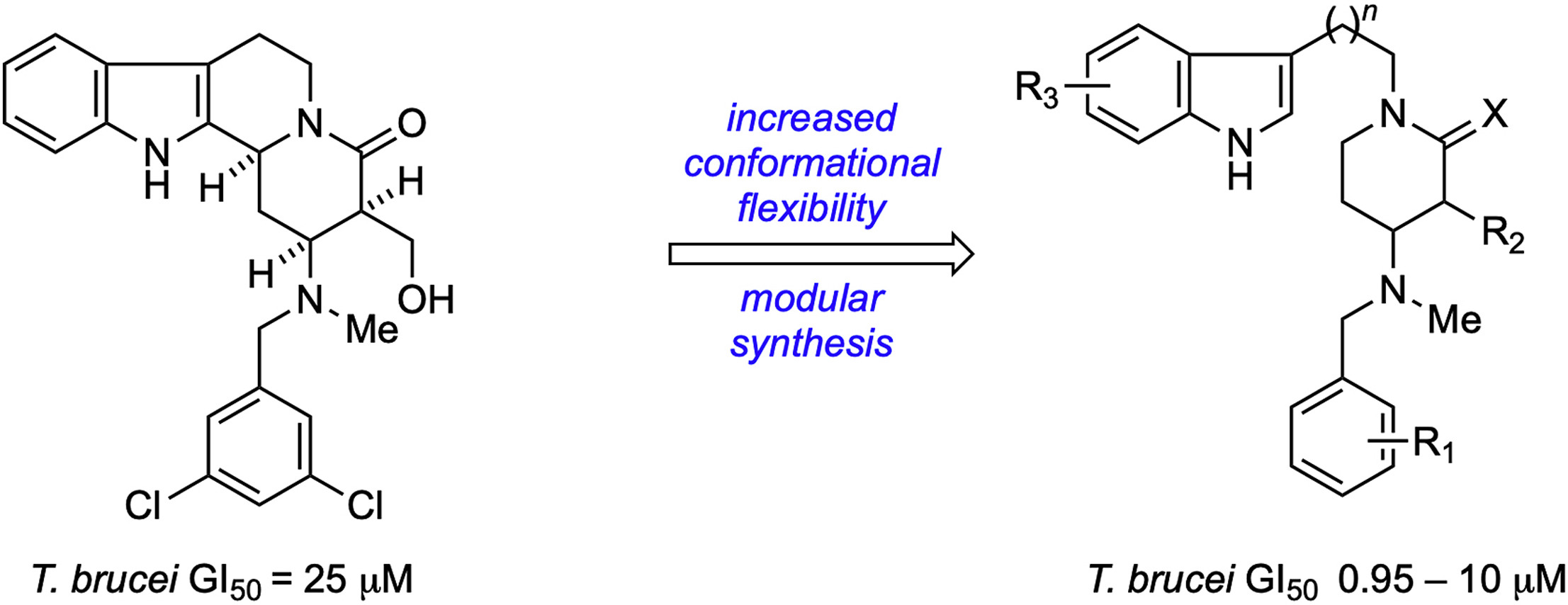
Chemical Optimization of CBL0137 for Human African Trypanosomiasis Lead Drug Discovery

The carbazole CBL0137 (1) is a lead for drug development against human African trypanosomiasis (HAT), a disease caused by Trypanosoma brucei. To advance 1 as a candidate drug, we synthesized new analogs that were evaluated for the physicochemical properties, antitrypanosome potency, selectivity against human cells, metabolism in microsomes or hepatocytes, and efflux ratios. Structure-activity/property analyses of analogs revealed …