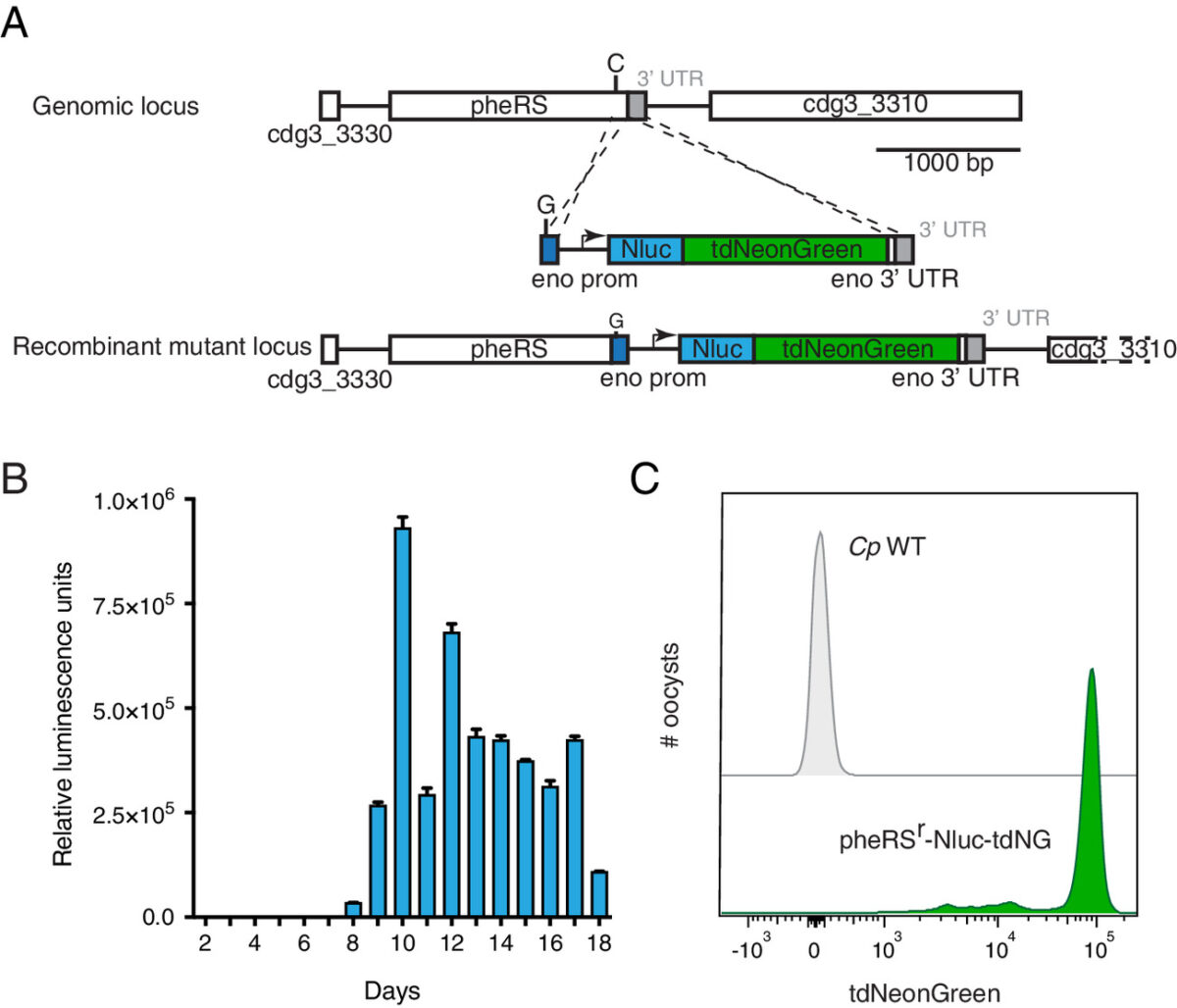
Genomic Characterization of Cryptosporidium spp. via iNextEra Library Preparation and Hybridization Bait Capture
Next-generation DNA sequencing (NGS) is used to study the genome sequences of Cryptosporidium spp., but NGS is challenging when pure Cryptosporidium oocysts are limited in number or not available. Varying levels of parasites present in fecal samples, combined with the abundance of host cells, bacterial and other microbial cells, and undigested food particles, often result …